









The da Vinci robot surgical system, manufactured by Intuitive Surgical, is a telesurgical 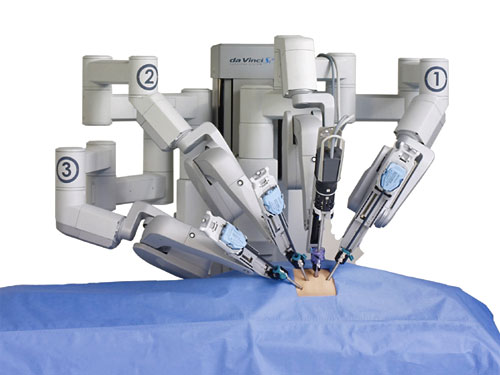 device that has been FDA approved for a range of minimally invasive procedures. During these procedures, the patient is on the operating table while the doctor sits several feet away at the control area of the machine, where he or she can maneuver the robotic arms while watching the procedure through a 3-D viewfinder. Many surgeons feel the robot makes it easier to see and navigate places in the body normally difficult to reach with traditional surgical methods. Doctors advocating the use of the da Vinci also point to the potential benefits for patients, such as small incisions, minimal bleeding, minimal scarring, and speeding post-surgery recovery time. As a result, surgical procedures using the da Vinci robot are becoming increasingly popular in the United States, with close to 400,000 of the procedures performed in the United States in 2012, and more than 1.5 million procedures since Intuitive Surgical received its initial FDA approval of the device.
device that has been FDA approved for a range of minimally invasive procedures. During these procedures, the patient is on the operating table while the doctor sits several feet away at the control area of the machine, where he or she can maneuver the robotic arms while watching the procedure through a 3-D viewfinder. Many surgeons feel the robot makes it easier to see and navigate places in the body normally difficult to reach with traditional surgical methods. Doctors advocating the use of the da Vinci also point to the potential benefits for patients, such as small incisions, minimal bleeding, minimal scarring, and speeding post-surgery recovery time. As a result, surgical procedures using the da Vinci robot are becoming increasingly popular in the United States, with close to 400,000 of the procedures performed in the United States in 2012, and more than 1.5 million procedures since Intuitive Surgical received its initial FDA approval of the device.
Unfortunately, as the da Vinci procedures have grown in popularity, so have the reports of injuries associated with their use. In the last 5 years, the U.S. Food and Drug administration has received more than 4,600 complaints from patients associated with robotic surgeries, including reports of serious burns, cuts, infections, including 89 deaths. Surgeries performed with the da Vinci have also been connected with damage to organs, harm to body tissue, and bleeding and internal scarring that can lead to chronic pain and dysfunction. Causing even greater concern is the fact many patients have been discharged from the hospital following a da Vinci robotic-assisted surgery only to discover the injury days later, thus requiring a prolonged hospitalization, and in some cases, additional surgery.
Many medical experts worry the high-tech, futuristic nature of the machine is driving surgeons’ decision to use the da Vinci, rather than an actual medical need, and they are not adequately considering the risks associate with its use. This fact, coupled with the lack of adequate training by surgeons using the device, has resulted in patients being subjected to surgical complications that may have been avoided if other safer surgical methods were chosen. Moreover, there is evidence the design of the da Vinci surgical robot is known to lead to injury even without any error on the part of the surgeon. As a result, there have been numerous, serious injuries attributed to the use of the da Vinci surgical robot, and lawsuits are being filed across the country against both Intuitive Surgical and negligent surgeons.
 A number of victims throughout the United States have filed lawsuits against Johnson & Johnson and its subsidiary, McNeil-PPC, after suffering severe liver damage, liver failure and, in some cases, death after taking Tylenol or Acetaminophen. The lawsuits allege that the Defendant manufacturers failed to provide adequate warnings about the risks associated with the side effects of Acetaminophen, which is the active ingredient in Tylenol. The victims suffered serious and life-threatening injuries, even when the medication was taken as directed. Liver damage can lead to prolonged hospitalizations and potentially, the need for a liver transplant. The average cost for a liver transplant in the United States exceeds $500,000.
A number of victims throughout the United States have filed lawsuits against Johnson & Johnson and its subsidiary, McNeil-PPC, after suffering severe liver damage, liver failure and, in some cases, death after taking Tylenol or Acetaminophen. The lawsuits allege that the Defendant manufacturers failed to provide adequate warnings about the risks associated with the side effects of Acetaminophen, which is the active ingredient in Tylenol. The victims suffered serious and life-threatening injuries, even when the medication was taken as directed. Liver damage can lead to prolonged hospitalizations and potentially, the need for a liver transplant. The average cost for a liver transplant in the United States exceeds $500,000.