

 Elmiron, the brand-name for the drug pentosan polysulfate sodium, is used to treat interstitial cystitis, which is a painful chronic bladder condition. Unfortunately, researchers have now discovered a clear link between the use of Elmiron and a progressive eye disorder known as pigmentary maculopathy. Pigmentary maculopathy affects the eye’s pigment and can cause impaired vision and even blindness after long-term use of Elmiron.
Elmiron, the brand-name for the drug pentosan polysulfate sodium, is used to treat interstitial cystitis, which is a painful chronic bladder condition. Unfortunately, researchers have now discovered a clear link between the use of Elmiron and a progressive eye disorder known as pigmentary maculopathy. Pigmentary maculopathy affects the eye’s pigment and can cause impaired vision and even blindness after long-term use of Elmiron.
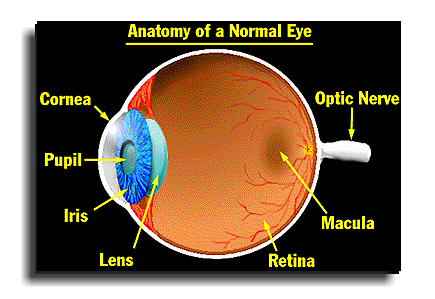
The correlation between Elmiron and pigmentary maculopathy was recently studied by researchers at the Emory University Medical School. The researchers observed that those treated for interstitial cystitis with Elmiron on a long-term protocol between 2015 and 2017 were at an increased risk of developing retinal maculopathy. These subjects reported significant difficulty reading and adapting in low-light situations. The researchers concluded that a connection exists between pigmentary maculopathy and chronic exposure to Elmiron that requires further research. They also urged patients who have taken Elmiron to be regularly screened by an ophthalmologist if they experience unusual vision symptoms and discontinue the drug if they are diagnosed with an eye disease.
The symptoms of pigmentary retinal maculopathy include: